ヒト常在菌を標的とした新たな治療・診断法開発への応用に期待
早稲田大学理工学術院の細川正人准教授、竹山春子教授、bitBiomeらの共同研究グループは13日、細菌叢から細菌ゲノムを個別取得するデータ解析フレームワーク「SMAGLinker」を開発したと発表した。
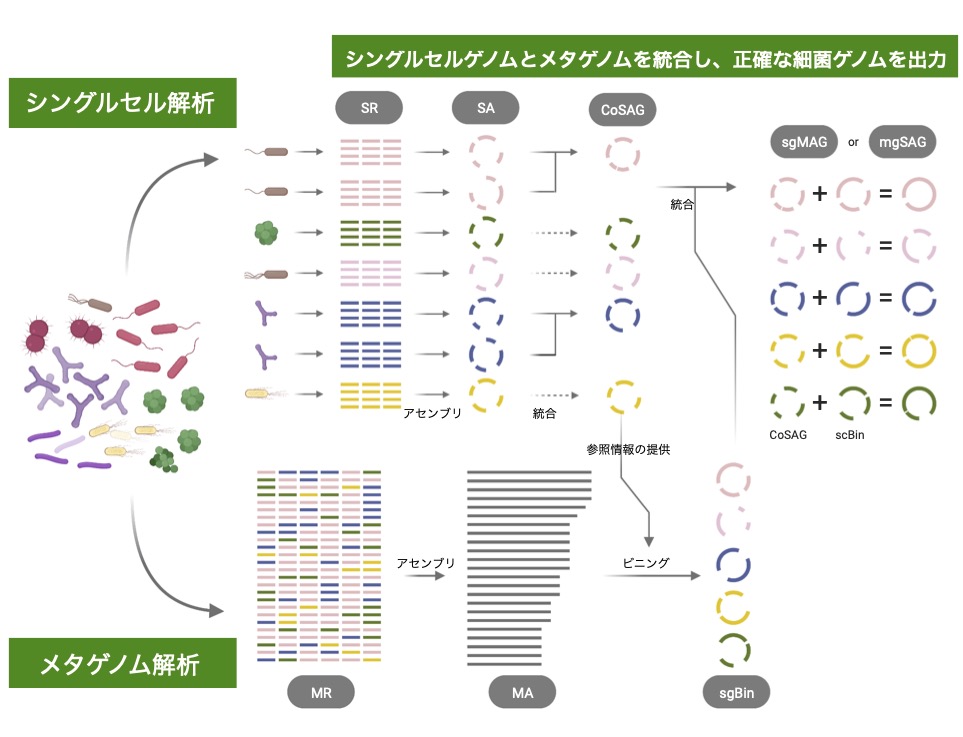
SMAGLinkerは、腸内細菌や皮膚常在菌などのヒト常在菌を単離培養することなく分析し、多様な細菌株のゲノムを正確かつ網羅的に獲得する「メタゲノム解析とシングルセル解析の統合データ解析法:シングルセルメタゲノミクス」を実現するためのフレームワーク。
同手法により、腸内細菌および皮膚常在菌の分析時に従来法より多種類・高品質な細菌ドラフトゲノムを取得でき、同種でわずかに異なる配列をもつ細菌株の識別が可能となった。
同手法を用いれば、個々人が持つヒト常在菌のゲノムを正確に決定でき、常在菌と宿主の相互作用の理解が進むことで、新たな治療・診断法の開発や常在菌由来物質の産業への応用が期待される。
細川氏は、早稲田大学関係ベンチャー bitBiomeを2018年に創業し、シングルセル解析技術の提供と研究開発を展開しており、 同研究成果は、Springer NatureグループのBioMed Central社が発刊するオープンアクセス科学誌『Microbiome』に12日に掲載された。
ヒト常在菌はヒトの健康と深く関わり、これらの細菌の理解は新たな医療・産業の創出に向け重要である。だが、細菌の多くは培養が難しく、その機能理解には多くの時間と労力が必要とされてきた。近年では、細菌を培養することなく、細菌群集からDNAを抽出して遺伝子配列を直接分析する「メタゲノム解析」が注目され、広く用いられるようになっている。
とはいえ、多量の遺伝子断片の集合であるメタゲノムから構築された細菌ゲノムは、一部配列の欠損や近縁細菌の配列の混入があり、正確性に欠けているため、似た配列をもつ細菌株の識別が困難であった。
そこで、細川氏らの研究グループは、従来のメタゲノム解析のデータを有効活用し、正確な細菌ゲノム情報の獲得を実現するために、シングルセル解析と統合したデータ解析法「シングルセルメタゲノミクス」を開発した。
シングルセル解析は同研究グループが以前より開発してきたもので、細胞1つずつ個別にゲノム配列を決定する特徴を持ち、近縁細菌・細菌株間の識別を可能とする。
だが、シングルセル解析単独では得られたゲノムが不完全であることが多い課題も残されていた。同手法では、ヒト常在菌サンプルからメタゲノム解析とシングルセル解析をそれぞれ実行してデータを得たのち、シングルセルデータを参照先としてメタゲノムデータの分別を行い、細菌ごとに配列を割り当てる。
最後に、ペアリングされた両データを統合することで、広範なメタゲノムデータと特異性の高いシングルセルデータの良さを足し合わせて、データを相互に補完した完全性の高いゲノム配列を出力します(図1)。
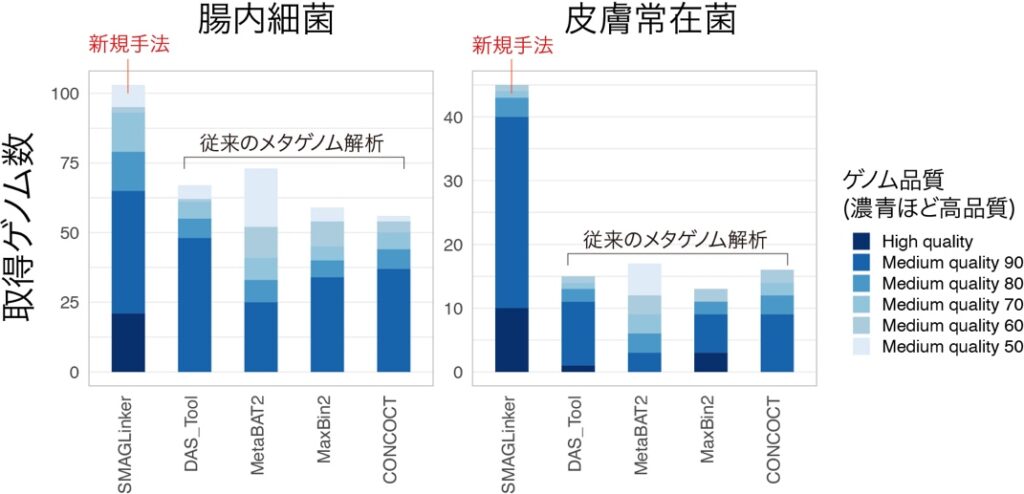
実際に、15種類の細菌を含む模擬サンプル、糞便由来腸内細菌、皮膚常在菌を対象とし、シングルセルメタゲノミクスの有効性を評価した。新規手法は従来法よりも正確な遺伝子配列の割当が実行されており、すべてのサンプルにおいて完全性が高い高品質な細菌ゲノムを数多く出力した(図2)。
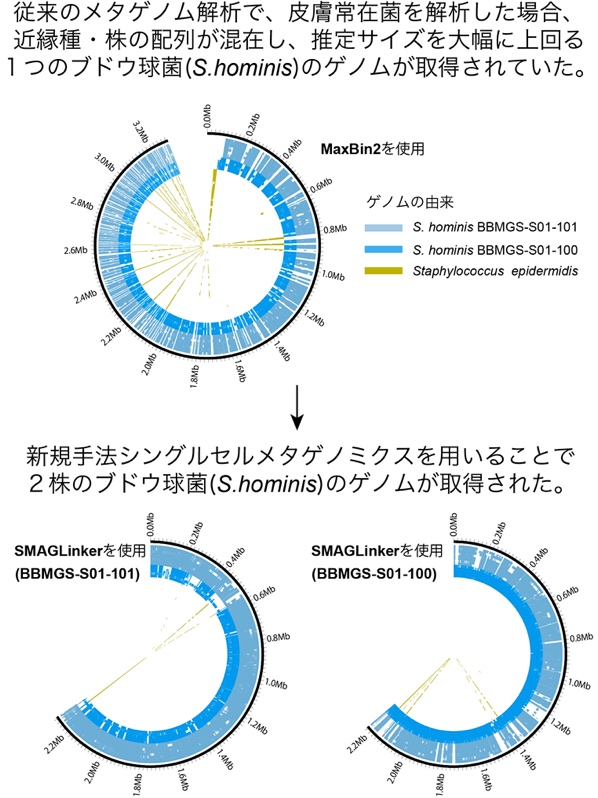
皮膚常在菌の解析では、メタゲノム単独解析では、ブドウ球菌(Staphylococcus hominis)のゲノムが1つ取得されたが、類縁菌の遺伝子配列が多く混入し、ゲノムサイズが推定値と大きく離れていたことから、ゲノム配列の誤った構築が行われたことが示唆された。
シングルセルメタゲノミクスを用いて取得データを再解析したところ、推定サイズと一致するゲノムが2つ取得され、測定サンプル中には皮膚常在菌として2種類のブドウ球菌株の存在が明らかになった。また、各細菌株が固有のプラスミドを有し、異なる形質を持つことが示唆された(図3)。
現在、ヒト常在菌の研究が世界中で活発に進んでいるが、同じ細菌種であっても外部からの遺伝子の取り込みや変異により異なる形質を持つことが明らかになっている。
同研究で開発したシングルセルメタゲノミクスにより、ヒト常在菌ゲノムが正確に決定されることで、個々人によって異なる細菌株の特性を正しく理解し、ヒトとの相互作用による健康や疾患との関わりについての理解の推進が期待される。
同研究成果は、bitBiomeにおいて、ヒト常在菌を標的とした新たな治療・診断法の開発や、常在菌が持つ酵素の化学工学・食品産業などへの利用に向けた研究開発の推進に活用される。
今後の課題としては、ヒト常在菌に比べて多種多様な細菌が存在する土壌細菌などの分析時には、メタゲノム配列の割当のために、より多くのシングルセルゲノム情報を必要とすることが挙げられる。
こうしたサンプルにも適用していくため、大規模なシングルセル解析を実行可能な技術開発を継続して行う。
◆細川氏のコメント
培養という従来の細菌叢研究に必須のプロセスをスキップして、全ゲノムを網羅的に獲得できることにより、そのサンプルの中で「誰が何をしているのか?」を予測することができる。
正確なデータの迅速な取得により、細菌叢研究のスピードや戦略性を革新したデータ駆動型の応用研究アプローチが実現できると考えている。

